D62 Preprocessing_Countdata
AGC, AY
15 3 2021
Last updated: 2023-08-29
Checks: 7 0
Knit directory: DEPDC5_D62_Analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220808) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 9dd12f1. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/02_D62_Preprocessing_counts.Rmd) and HTML
(docs/02_D62_Preprocessing_counts.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 9dd12f1 | achiocch | 2023-08-29 | wflow_publish(c("./analysis/", "code/"), all = T) |
| html | 20e7956 | achiocch | 2023-07-31 | Build site. |
| Rmd | fbbab8e | achiocch | 2023-07-31 | wflow_publish(c("./analysis/", "code/"), all = T) |
| Rmd | 6cbb9a0 | achiocch | 2023-07-25 | plots fro publication added |
| Rmd | c1f2468 | achiocch | 2022-10-11 | sets the installation procedure in the readme |
| html | c1f2468 | achiocch | 2022-10-11 | sets the installation procedure in the readme |
| Rmd | a7c5f57 | achiocch | 2022-10-10 | fix intaller |
| html | a7c5f57 | achiocch | 2022-10-10 | fix intaller |
| Rmd | 59a7df2 | achiocch | 2022-09-22 | minor changes |
| Rmd | dc78d32 | Andreas Geburtig-Chiocchetti | 2022-08-09 | full analysis pre manuscript version |
| html | dc78d32 | Andreas Geburtig-Chiocchetti | 2022-08-09 | full analysis pre manuscript version |
| Rmd | f249225 | achiocch | 2022-08-08 | adds data and code |
Preprocessing
home = getwd()
output= paste0(home, "/output/")
source(paste0(home,"/code/custom_functions.R")) #also defines colorsLade nötiges Paket: kableExtra
Attache Paket: 'kableExtra'Das folgende Objekt ist maskiert 'package:dplyr':
group_rowsLade nötiges Paket: compareGroupsfiletarget= paste0(home,"/data/Countmatrix.RData")
load(filetarget)
Ntot= nrow(Countdata)
#merge non unique annotations
if(length(unique(rownames(Countdata))) != Ntot){
Countdata = Countdata %>% group_by(row.names(Countdata)) %>% summarise_each(sum)
Ntot= nrow(Countdata)
}
hgnc=gconvert(query=as.numeric(rownames(Countdata)),
organism = "hsapiens",
numeric_ns = "ENTREZGENE_ACC",
target = "HGNC")
Ids = hgnc %>% dplyr::select(name, input, description) %>% group_by(input) %>%
summarise(name=paste(name, sep="; ", collapse = ";"), description = dplyr::first(description))
rowdescription = data.frame(entrez_gene = Ids$input,
hgnc=Ids$name,
description=Ids$description)
rowdescription = rowdescription[match(row.names(Countdata), rowdescription$entrez_gene),]
rownames(rowdescription)=row.names(Countdata)
# load and parse sample information
SampleInfo=read.csv2(paste0(home,"/data/D62_Sample_info_CePTER_RNASeq.csv"),
row.names = 1)
SampleInfo$Row=gsub("[0-9]*","",SampleInfo$Position)
SampleInfo$Col=as.numeric(gsub("[A-Z]*","",SampleInfo$Position))
# set factors and relevel
SampleInfo$CellLine = as.factor(SampleInfo$CellLine)
SampleInfo$gRNA = paste0("sg",SampleInfo$gRNA)
SampleInfo$gRNA = factor(SampleInfo$gRNA, levels=c("sgNTC", "sg2.1", "sg2.2"),
labels=c("sgNTC", "sg2.1", "sg2.2"))
SampleInfo$gRNA = relevel(SampleInfo$gRNA,ref="sgNTC" )
SampleInfo$KO = factor(SampleInfo$KO, levels=c(T,F), labels=c("KO", "WT"))
SampleInfo$KO = relevel(SampleInfo$KO,ref="WT" )
SampleInfo$DIFF = factor(SampleInfo$DIFF, levels=c(TRUE,FALSE),
labels=c("DIFF", "noDIFF"))
SampleInfo$DIFF = relevel(SampleInfo$DIFF,ref="noDIFF")
SampleInfo$RAPA = factor(SampleInfo$RAPA, levels=c(T,F),
labels=c("RAPA", "noRAPA"))
SampleInfo$RAPA = relevel(SampleInfo$RAPA,ref="noRAPA")
SampleInfo$label = with(SampleInfo, paste(CellLine,gRNA,DIFF,RAPA, sep="_"))
SampleInfo$fastQID = rownames(SampleInfo)
SampleInfo = SampleInfo %>% dplyr::group_by(label) %>% mutate(replicate=seq(n())) %>% as.data.frame()
SampleInfo$label_rep=with(SampleInfo, paste(label,replicate,sep="_"))
rownames(SampleInfo)=SampleInfo$fastQID
# align datasets
checkfiles = all(rownames(SampleInfo) %in% colnames(Countdata))
IDs=intersect(rownames(SampleInfo), colnames(Countdata))
Countdata = Countdata[,IDs]
SampleInfo = SampleInfo[IDs, ]
SampleInfo$reads_per_sample = colSums(Countdata)display_tab(head(Countdata))| DE10NGSUKBR112901 | DE80NGSUKBR112902 | DE53NGSUKBR112903 | DE26NGSUKBR112904 | DE96NGSUKBR112905 | DE69NGSUKBR112906 | DE42NGSUKBR112907 | DE15NGSUKBR112908 | DE85NGSUKBR112909 | DE58NGSUKBR112910 | DE31NGSUKBR112911 | DE04NGSUKBR112912 | DE74NGSUKBR112913 | DE47NGSUKBR112914 | DE20NGSUKBR112915 | DE90NGSUKBR112916 | DE63NGSUKBR112917 | DE36NGSUKBR112918 | DE09NGSUKBR112919 | DE79NGSUKBR112920 | DE52NGSUKBR112921 | DE25NGSUKBR112922 | DE95NGSUKBR112923 | DE68NGSUKBR112924 | DE41NGSUKBR112925 | DE14NGSUKBR112926 | DE84NGSUKBR112927 | DE57NGSUKBR112928 | DE30NGSUKBR112929 | DE03NGSUKBR112930 | DE73NGSUKBR112931 | DE46NGSUKBR112932 | DE19NGSUKBR112933 | DE89NGSUKBR112934 | DE62NGSUKBR112935 | DE35NGSUKBR112936 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 100287102 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 653635 | 46 | 82 | 133 | 121 | 84 | 78 | 31 | 21 | 32 | 48 | 26 | 47 | 59 | 18 | 113 | 66 | 71 | 99 | 101 | 59 | 55 | 75 | 77 | 42 | 27 | 32 | 41 | 85 | 90 | 112 | 75 | 0 | 32 | 25 | 42 | 19 |
| 102466751 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 100302278 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 645520 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 79501 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
display_tab(SampleInfo)| Plate | Position | Row | Col | CellLine | gRNA | KO | DIFF | RAPA | Conc | UV260_280 | UV260_230 | label | fastQID | replicate | label_rep | reads_per_sample | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DE10NGSUKBR112901 | 1 | A01 | A | 1 | D62 | sgNTC | WT | DIFF | RAPA | 4 | 2.5 | 0.588 | D62_sgNTC_DIFF_RAPA | DE10NGSUKBR112901 | 1 | D62_sgNTC_DIFF_RAPA_1 | 8167558 |
| DE80NGSUKBR112902 | 1 | A02 | A | 2 | D62 | sgNTC | WT | DIFF | RAPA | 3.6 | 1.8 | 0.529 | D62_sgNTC_DIFF_RAPA | DE80NGSUKBR112902 | 2 | D62_sgNTC_DIFF_RAPA_2 | 7947513 |
| DE53NGSUKBR112903 | 1 | A03 | A | 3 | D62 | sgNTC | WT | DIFF | RAPA | 4 | 2 | 0.714 | D62_sgNTC_DIFF_RAPA | DE53NGSUKBR112903 | 3 | D62_sgNTC_DIFF_RAPA_3 | 8927353 |
| DE26NGSUKBR112904 | 1 | A04 | A | 4 | D62 | sgNTC | WT | DIFF | noRAPA | 14.8 | 1.682 | 1.276 | D62_sgNTC_DIFF_noRAPA | DE26NGSUKBR112904 | 1 | D62_sgNTC_DIFF_noRAPA_1 | 6192682 |
| DE96NGSUKBR112905 | 1 | A05 | A | 5 | D62 | sgNTC | WT | DIFF | noRAPA | 10.4 | 2.167 | 1.529 | D62_sgNTC_DIFF_noRAPA | DE96NGSUKBR112905 | 2 | D62_sgNTC_DIFF_noRAPA_2 | 6316070 |
| DE69NGSUKBR112906 | 1 | A06 | A | 6 | D62 | sgNTC | WT | DIFF | noRAPA | 8 | 2.22 | 1.33 | D62_sgNTC_DIFF_noRAPA | DE69NGSUKBR112906 | 3 | D62_sgNTC_DIFF_noRAPA_3 | 7211176 |
| DE42NGSUKBR112907 | 1 | A07 | A | 7 | D62 | sg2.1 | KO | DIFF | RAPA | 4 | 2.5 | 1.25 | D62_sg2.1_DIFF_RAPA | DE42NGSUKBR112907 | 1 | D62_sg2.1_DIFF_RAPA_1 | 6472088 |
| DE15NGSUKBR112908 | 1 | A08 | A | 8 | D62 | sg2.1 | KO | DIFF | RAPA | 5.2 | 1.857 | 1.444 | D62_sg2.1_DIFF_RAPA | DE15NGSUKBR112908 | 2 | D62_sg2.1_DIFF_RAPA_2 | 6381728 |
| DE85NGSUKBR112909 | 1 | A09 | A | 9 | D62 | sg2.1 | KO | DIFF | RAPA | 5.6 | 2 | 1.556 | D62_sg2.1_DIFF_RAPA | DE85NGSUKBR112909 | 3 | D62_sg2.1_DIFF_RAPA_3 | 7515594 |
| DE58NGSUKBR112910 | 1 | A10 | A | 10 | D62 | sg2.1 | KO | DIFF | noRAPA | 12.4 | 1.938 | 1.722 | D62_sg2.1_DIFF_noRAPA | DE58NGSUKBR112910 | 1 | D62_sg2.1_DIFF_noRAPA_1 | 8072060 |
| DE31NGSUKBR112911 | 1 | A11 | A | 11 | D62 | sg2.1 | KO | DIFF | noRAPA | 13.6 | 2 | 0.895 | D62_sg2.1_DIFF_noRAPA | DE31NGSUKBR112911 | 2 | D62_sg2.1_DIFF_noRAPA_2 | 9132042 |
| DE04NGSUKBR112912 | 1 | A12 | A | 12 | D62 | sg2.1 | KO | DIFF | noRAPA | 8.4 | 1.909 | 0.244 | D62_sg2.1_DIFF_noRAPA | DE04NGSUKBR112912 | 3 | D62_sg2.1_DIFF_noRAPA_3 | 9158749 |
| DE74NGSUKBR112913 | 1 | B01 | B | 1 | D62 | sg2.2 | KO | DIFF | RAPA | 4.4 | 1.833 | 1.571 | D62_sg2.2_DIFF_RAPA | DE74NGSUKBR112913 | 1 | D62_sg2.2_DIFF_RAPA_1 | 8022580 |
| DE47NGSUKBR112914 | 1 | B02 | B | 2 | D62 | sg2.2 | KO | DIFF | RAPA | 4.4 | 1.833 | 0.5 | D62_sg2.2_DIFF_RAPA | DE47NGSUKBR112914 | 2 | D62_sg2.2_DIFF_RAPA_2 | 5290014 |
| DE20NGSUKBR112915 | 1 | B03 | B | 3 | D62 | sg2.2 | KO | DIFF | RAPA | 6 | 1.875 | 0.172 | D62_sg2.2_DIFF_RAPA | DE20NGSUKBR112915 | 3 | D62_sg2.2_DIFF_RAPA_3 | 7350899 |
| DE90NGSUKBR112916 | 1 | B04 | B | 4 | D62 | sg2.2 | KO | DIFF | noRAPA | 5.2 | 1.857 | 0.334 | D62_sg2.2_DIFF_noRAPA | DE90NGSUKBR112916 | 1 | D62_sg2.2_DIFF_noRAPA_1 | 7142950 |
| DE63NGSUKBR112917 | 1 | B05 | B | 5 | D62 | sg2.2 | KO | DIFF | noRAPA | 6 | 1.667 | 0.789 | D62_sg2.2_DIFF_noRAPA | DE63NGSUKBR112917 | 2 | D62_sg2.2_DIFF_noRAPA_2 | 6379496 |
| DE36NGSUKBR112918 | 1 | B06 | B | 6 | D62 | sg2.2 | KO | DIFF | noRAPA | 4 | 1.667 | 1.25 | D62_sg2.2_DIFF_noRAPA | DE36NGSUKBR112918 | 3 | D62_sg2.2_DIFF_noRAPA_3 | 6345821 |
| DE09NGSUKBR112919 | 1 | B07 | B | 7 | D62 | sgNTC | WT | noDIFF | RAPA | 22 | 2.037 | 1.25 | D62_sgNTC_noDIFF_RAPA | DE09NGSUKBR112919 | 1 | D62_sgNTC_noDIFF_RAPA_1 | 6954777 |
| DE79NGSUKBR112920 | 1 | B08 | B | 8 | D62 | sgNTC | WT | noDIFF | RAPA | 14.8 | 2.176 | 0.698 | D62_sgNTC_noDIFF_RAPA | DE79NGSUKBR112920 | 2 | D62_sgNTC_noDIFF_RAPA_2 | 6247879 |
| DE52NGSUKBR112921 | 1 | B09 | B | 9 | D62 | sgNTC | WT | noDIFF | RAPA | 19.2 | 2.087 | 1.371 | D62_sgNTC_noDIFF_RAPA | DE52NGSUKBR112921 | 3 | D62_sgNTC_noDIFF_RAPA_3 | 7651123 |
| DE25NGSUKBR112922 | 1 | B10 | B | 10 | D62 | sgNTC | WT | noDIFF | noRAPA | 16.8 | 1.909 | 0.525 | D62_sgNTC_noDIFF_noRAPA | DE25NGSUKBR112922 | 1 | D62_sgNTC_noDIFF_noRAPA_1 | 8143934 |
| DE95NGSUKBR112923 | 1 | B11 | B | 11 | D62 | sgNTC | WT | noDIFF | noRAPA | 18.8 | 1.958 | 1.343 | D62_sgNTC_noDIFF_noRAPA | DE95NGSUKBR112923 | 2 | D62_sgNTC_noDIFF_noRAPA_2 | 7710977 |
| DE68NGSUKBR112924 | 1 | B12 | B | 12 | D62 | sgNTC | WT | noDIFF | noRAPA | 17.6 | 2 | 1.189 | D62_sgNTC_noDIFF_noRAPA | DE68NGSUKBR112924 | 3 | D62_sgNTC_noDIFF_noRAPA_3 | 9158766 |
| DE41NGSUKBR112925 | 1 | C01 | C | 1 | D62 | sg2.1 | KO | noDIFF | RAPA | 26.8 | 2.03 | 1.914 | D62_sg2.1_noDIFF_RAPA | DE41NGSUKBR112925 | 1 | D62_sg2.1_noDIFF_RAPA_1 | 7883862 |
| DE14NGSUKBR112926 | 1 | C02 | C | 2 | D62 | sg2.1 | KO | noDIFF | RAPA | 24.8 | 2.067 | 1.59 | D62_sg2.1_noDIFF_RAPA | DE14NGSUKBR112926 | 2 | D62_sg2.1_noDIFF_RAPA_2 | 7134773 |
| DE84NGSUKBR112927 | 1 | C03 | C | 3 | D62 | sg2.1 | KO | noDIFF | RAPA | 20.8 | 2.167 | 1.268 | D62_sg2.1_noDIFF_RAPA | DE84NGSUKBR112927 | 3 | D62_sg2.1_noDIFF_RAPA_3 | 8617208 |
| DE57NGSUKBR112928 | 1 | C04 | C | 4 | D62 | sg2.1 | KO | noDIFF | noRAPA | 14.4 | 2.571 | 0.184 | D62_sg2.1_noDIFF_noRAPA | DE57NGSUKBR112928 | 1 | D62_sg2.1_noDIFF_noRAPA_1 | 7544453 |
| DE30NGSUKBR112929 | 1 | C05 | C | 5 | D62 | sg2.1 | KO | noDIFF | noRAPA | 16.4 | 2.158 | 0.911 | D62_sg2.1_noDIFF_noRAPA | DE30NGSUKBR112929 | 2 | D62_sg2.1_noDIFF_noRAPA_2 | 7622380 |
| DE03NGSUKBR112930 | 1 | C06 | C | 6 | D62 | sg2.1 | KO | noDIFF | noRAPA | 12.8 | 2.286 | 0.711 | D62_sg2.1_noDIFF_noRAPA | DE03NGSUKBR112930 | 3 | D62_sg2.1_noDIFF_noRAPA_3 | 7939374 |
| DE73NGSUKBR112931 | 1 | C07 | C | 7 | D62 | sg2.2 | KO | noDIFF | RAPA | 19.6 | 1.96 | 1.69 | D62_sg2.2_noDIFF_RAPA | DE73NGSUKBR112931 | 1 | D62_sg2.2_noDIFF_RAPA_1 | 7320431 |
| DE46NGSUKBR112932 | 1 | C08 | C | 8 | D62 | sg2.2 | KO | noDIFF | RAPA | 18 | 2.045 | 1.607 | D62_sg2.2_noDIFF_RAPA | DE46NGSUKBR112932 | 2 | D62_sg2.2_noDIFF_RAPA_2 | 6532522 |
| DE19NGSUKBR112933 | 1 | C09 | C | 9 | D62 | sg2.2 | KO | noDIFF | RAPA | 17.6 | 2 | 1.63 | D62_sg2.2_noDIFF_RAPA | DE19NGSUKBR112933 | 3 | D62_sg2.2_noDIFF_RAPA_3 | 7115292 |
| DE89NGSUKBR112934 | 1 | C10 | C | 10 | D62 | sg2.2 | KO | noDIFF | noRAPA | 15.2 | 2.111 | 1.583 | D62_sg2.2_noDIFF_noRAPA | DE89NGSUKBR112934 | 1 | D62_sg2.2_noDIFF_noRAPA_1 | 7618310 |
| DE62NGSUKBR112935 | 1 | C11 | C | 11 | D62 | sg2.2 | KO | noDIFF | noRAPA | 14.4 | 2.25 | 1.091 | D62_sg2.2_noDIFF_noRAPA | DE62NGSUKBR112935 | 2 | D62_sg2.2_noDIFF_noRAPA_2 | 7387684 |
| DE35NGSUKBR112936 | 1 | C12 | C | 12 | D62 | sg2.2 | KO | noDIFF | noRAPA | 15.2 | 2.111 | 0.717 | D62_sg2.2_noDIFF_noRAPA | DE35NGSUKBR112936 | 3 | D62_sg2.2_noDIFF_noRAPA_3 | 7440730 |
Total number of samples overlapping between Counts and SampleInfo: 36
boxplot_counts = function(plotsubset,maintitle,colorcode){
vals=log2(plotsubset+1)
a =boxplot(vals, main = maintitle,
col = Dark8[as.factor(SampleInfo[,colorcode])], names=NA,
ylab = "log2 transformed", xlab="samples", xaxt="n")
legend(ncol(vals)*1.1, max(vals), legend = levels(SampleInfo[,colorcode]),
bg="white",xpd=T,box.col = "white",
pch = 16, col = Dark8[1:length(unique(SampleInfo[,colorcode]))])
}
barplot_counts = function(DF, maintitle, colorcode) {
vals=log2(DF[,"reads_per_sample"])
barplot(vals, main = maintitle,
col = Dark8[as.factor(DF[,colorcode])], names=NA, xaxt="n",
ylab = "log2 transformed", xlab="samples")
legend(length(vals)*1.25, max(vals), legend = levels(DF[,colorcode]), pch = 16,
bg ="white",xpd=T, box.col="white",
col = Dark8[1:length(unique(DF[,colorcode]))])
}
par(mar=c(3,5,5,7))
boxplot_counts(Countdata, "raw counts", "gRNA")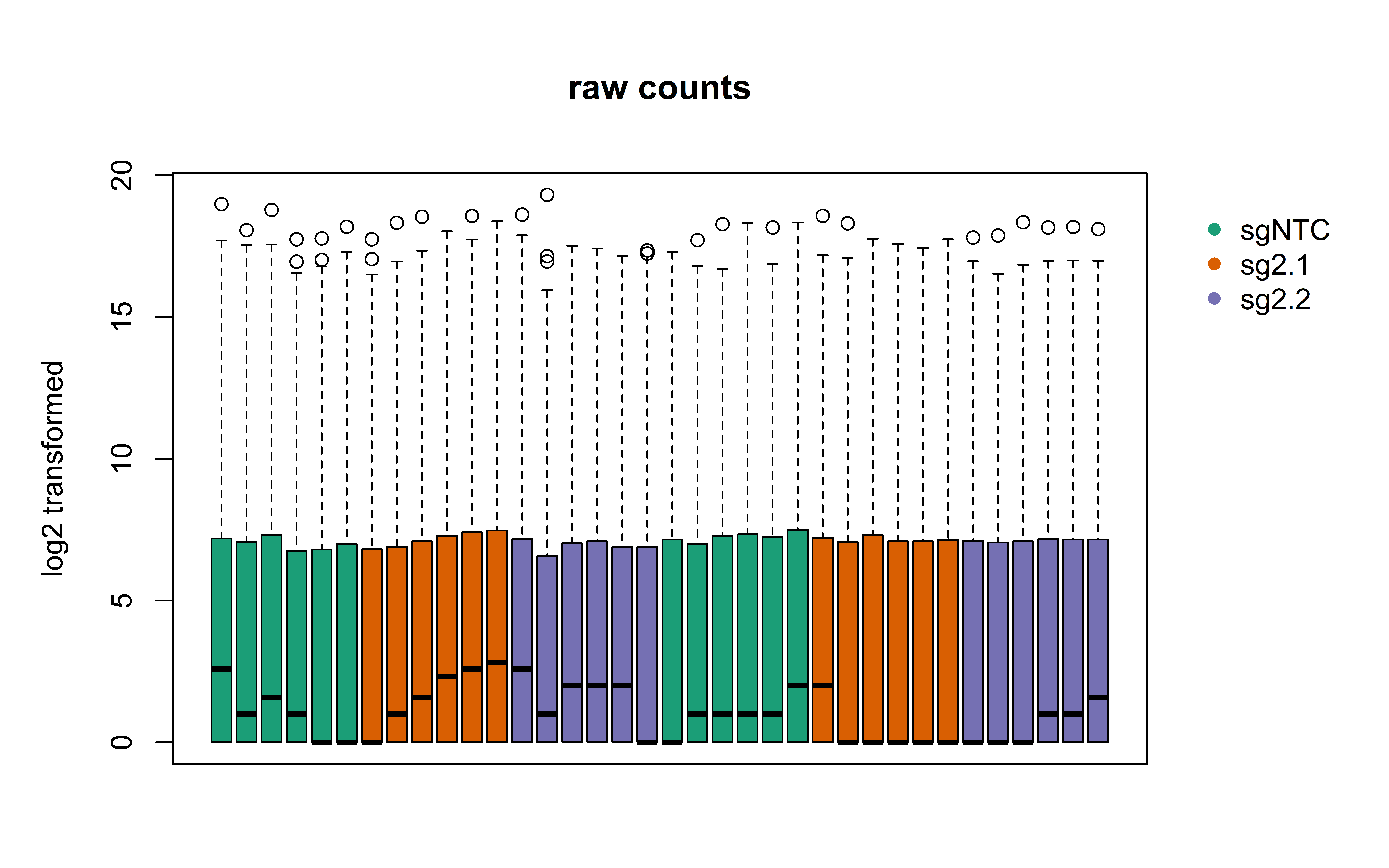
barplot_counts(SampleInfo, "total reads", "gRNA")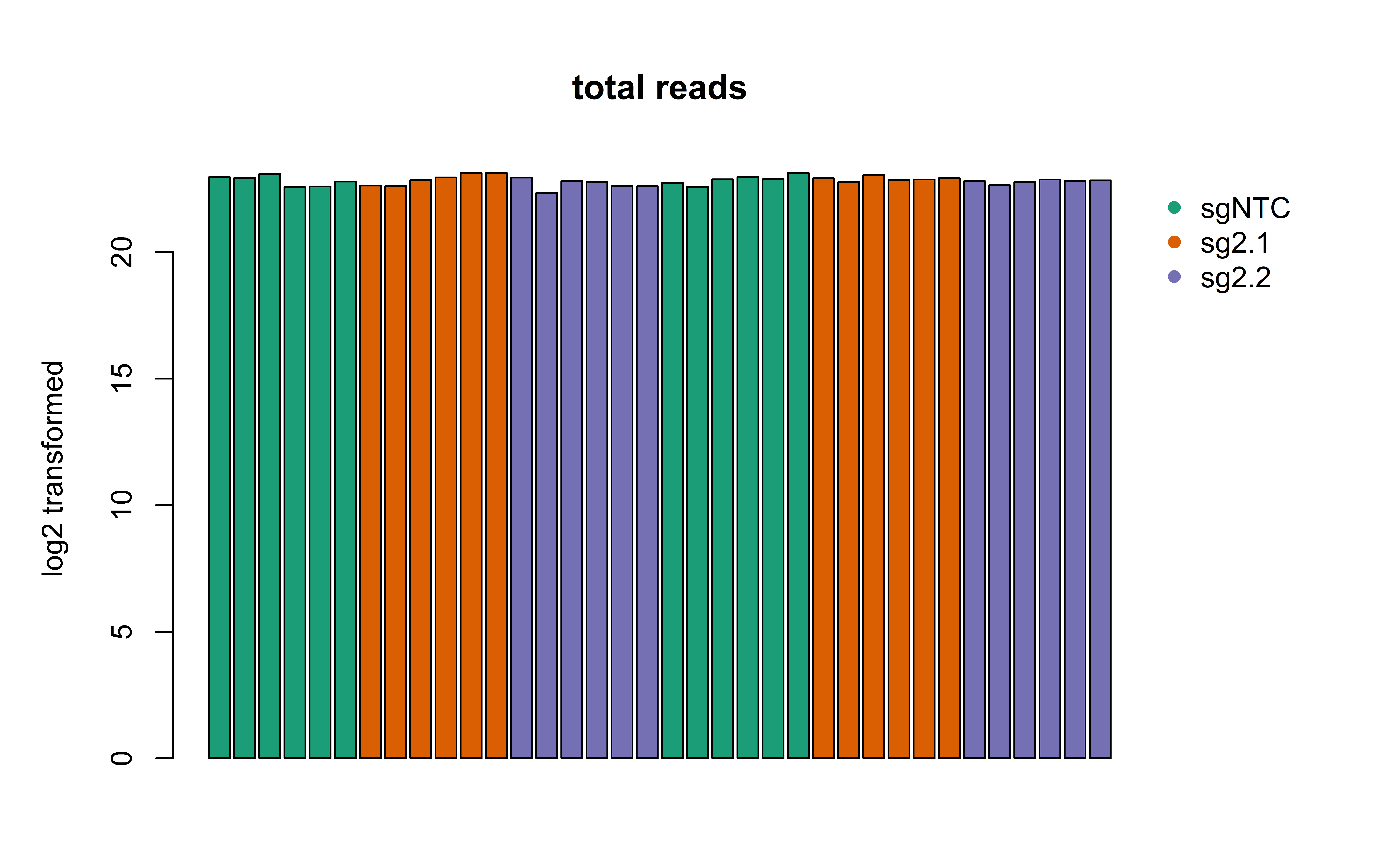
plot(density(log2(rowMeans(Countdata))), main="distribution of gene expression",
xlab="mean log2(counts +1)")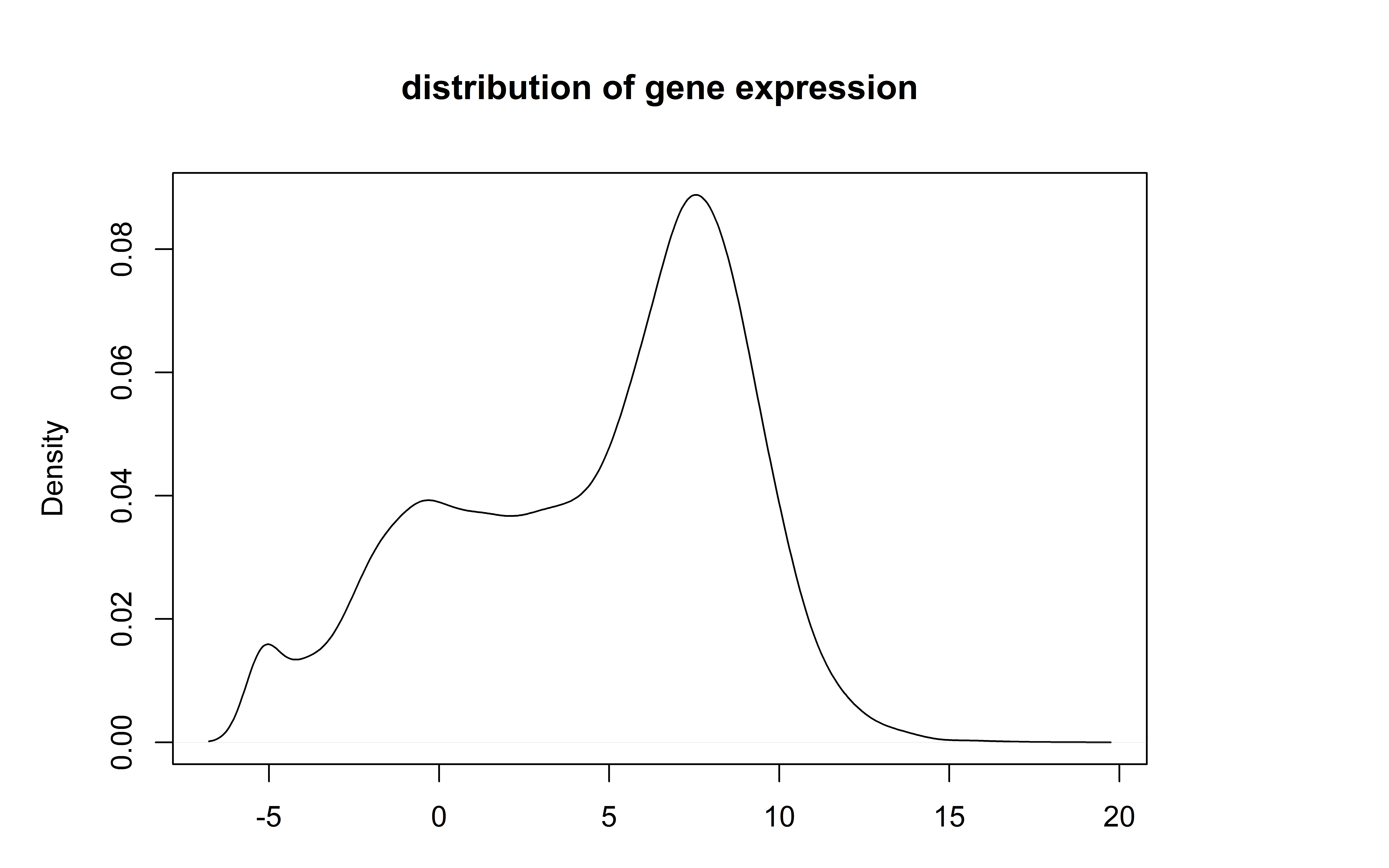
# remove genes wich were not detected in at least 50% of the samples
keeperidx = rowSums(Countdata>1)>nrow(SampleInfo)/2
Countdata_cl = Countdata[keeperidx, ]
rowdescription = rowdescription[row.names(Countdata_cl),]
fullmodel = as.formula("~gRNA+DIFF+RAPA")
ddsMat <- DESeqDataSetFromMatrix(countData = Countdata_cl,
colData = SampleInfo,
rowData = rowdescription,
design = fullmodel)
ddsMat = estimateSizeFactors(ddsMat)
ddsMat = estimateDispersions(ddsMat)gene-wise dispersion estimatesmean-dispersion relationshipfinal dispersion estimatesreads = as.data.frame(counts(ddsMat, normalized=T))
SDs = apply(reads, 1, sd)
keepvar = SDs>0
ddsMat <- ddsMat[keepvar,]
Nfilt = length(ddsMat)
reads = as.data.frame(counts(ddsMat, normalized=T))
SampleInfo$reads_per_sample_cl= colSums(reads)before cleaning
- Average reads per samples: 7 445 857
- Standard deviation reads per samples: 921 177.8
- Total genes mapped: 28 395
after cleaning
- Average reads per samples: 7 195 819
- Standard deviation reads per samples: 345 374
- Genes removed due to low reads: 14 664
- Total genes included after filtering: 13 731
Clustering
hierarchical clustering based on the top 2000 genes by variance
log2_cpm = log2(reads+1)
varsset=apply(log2_cpm, 1, var)
cpm.sel.trans = t(log2_cpm[order(varsset,decreasing = T)[1:2000],])
rownames(cpm.sel.trans)=SampleInfo$label_rep
distance = dist(cpm.sel.trans)
hc = stats::hclust(distance, method="ward.D2")
cutN=12 #number of different conditions (DIFF, RAPA, sgRNA)
clusters = cutree(hc, k=cutN)
Colors=sample(jetcolors(cutN))[clusters]
myLetters <- LETTERS[1:26]
numRow=match(SampleInfo$Row, myLetters)
numRow=numRow+(SampleInfo$Plate-1)*8
addRow=LETTERS[numRow]
Plotdata=data.frame(Rows=addRow, numRow = numRow, Cols = SampleInfo$Col,
Group=clusters, Colors=Colors)
par(mar=c(15,3,5,3))
plot(as.dendrogram(hc), main=paste("Similairtiy by gene expression, guessed",cutN,"clusters"), cex=0.7)
colored_dots(colors = Colors, dend = as.dendrogram(hc), rowLabels = "cluster")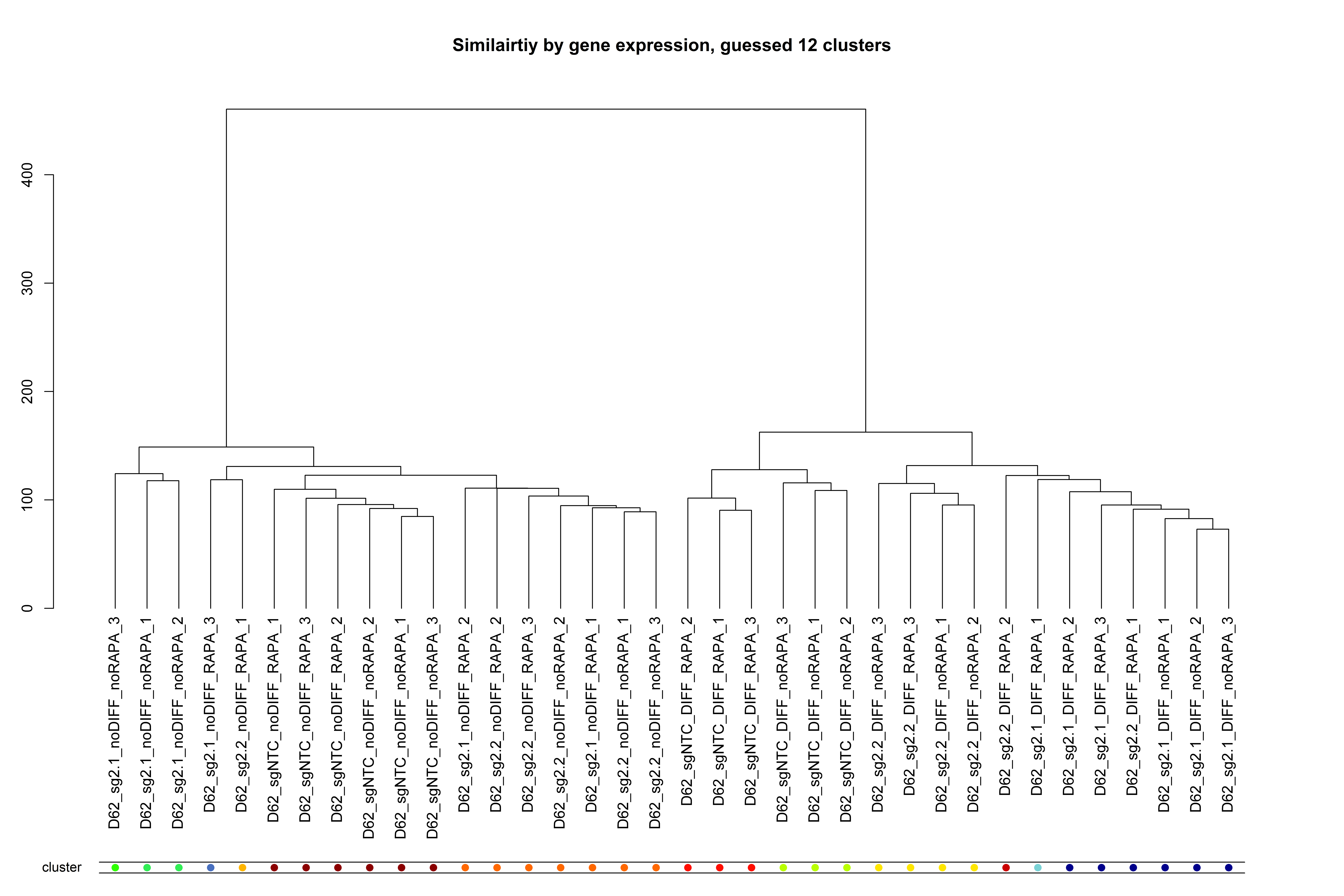
Similarity based on hcluster plot
par(mar=c(2,5,8,3))
plot(0,0, type="n", ylab="", xlab="",
ylim=rev(range(Plotdata$numRow))+c(1,-1),
xlim=range(Plotdata$Cols)+c(-1,1), xaxt="n",yaxt="n" ,
main="plate similarity plot")
points(y=Plotdata$numRow, x=Plotdata$Cols, pch=16, cex=4, col=Plotdata$Colors)
text(y=Plotdata$numRow, x=Plotdata$Cols, labels = Plotdata$Group)
text(y=Plotdata$numRow, x=Plotdata$Cols, labels = Plotdata$Group)
axis(2, at=1:9, labels = c(paste0("P1_", LETTERS[1:8]), "P2_A"), las=1)
axis(3, at=1:12, labels = c(paste0("Col_", 1:12)), las=3)
abline(h=8.5)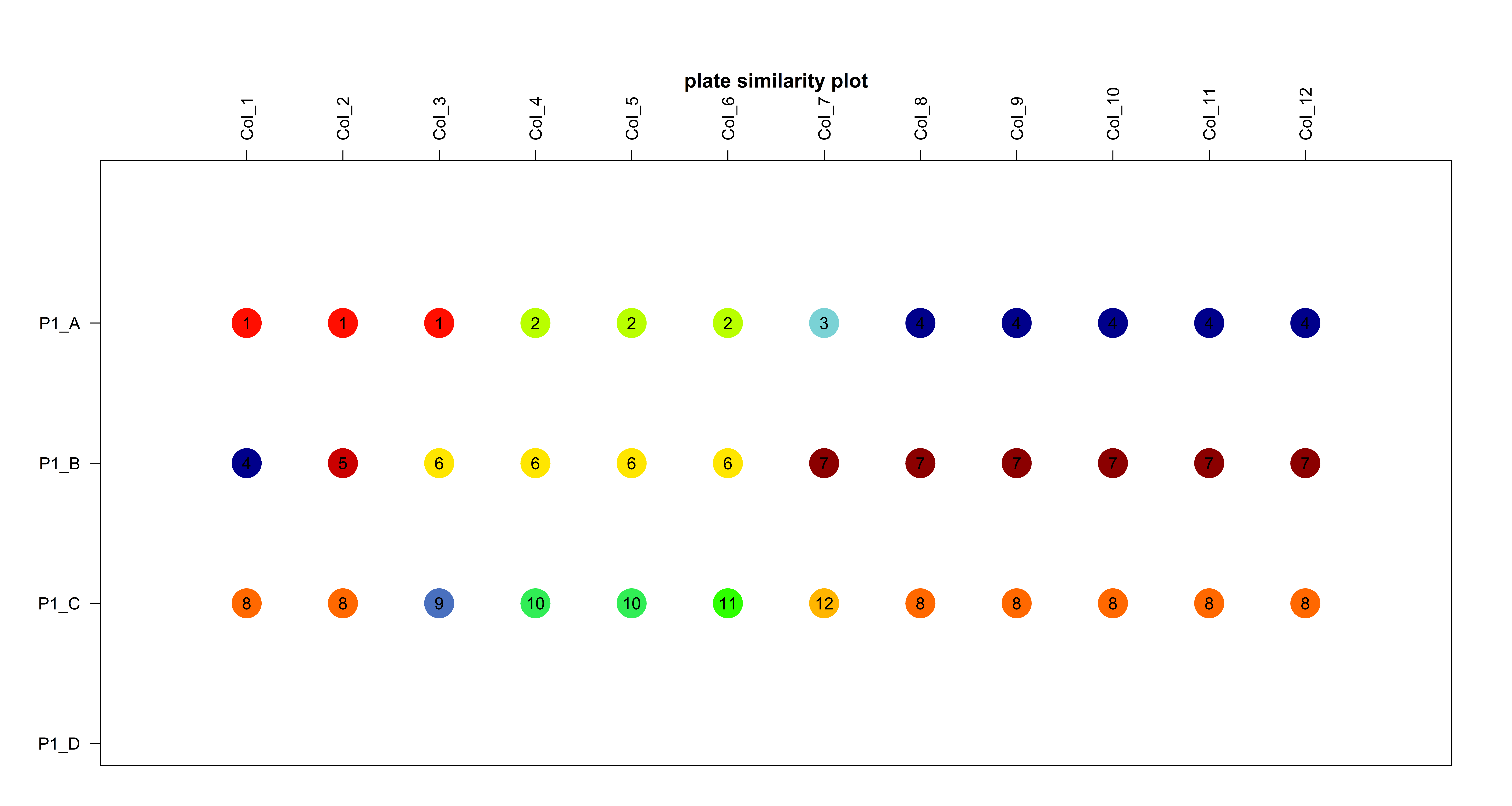
sampleDistMatrix <- as.matrix(distance)
#colors for plotting heatmap
colors <- colorRampPalette(brewer.pal(9, "Spectral"))(255)
cellcol = Dark8[1:nlevels(SampleInfo$CellLine)]
names(cellcol) = levels(SampleInfo$CellLine)
gRNAcol = Dark8[c(1:nlevels(SampleInfo$gRNA))+nlevels(SampleInfo$CellLine)]
names(gRNAcol) = levels(SampleInfo$gRNA)
diffcol = brewer.pal(3,"Set1")[1:nlevels(SampleInfo$DIFF)]
names(diffcol) = levels(SampleInfo$DIFF)
rapacol = brewer.pal(3,"Set2")[1:nlevels(SampleInfo$RAPA)]
names(rapacol) = levels(SampleInfo$RAPA)
ann_colors = list(
DIFF = diffcol,
RAPA = rapacol,
gRNA = gRNAcol
#,CellLine=cellcol
)
labels = SampleInfo[,c("gRNA","DIFF", "RAPA")] %>%
mutate_all(as.character) %>% as.data.frame()
rownames(labels)=SampleInfo$label_rep
pheatmap(sampleDistMatrix,
clustering_distance_rows = distance,
clustering_distance_cols = distance,
clustering_method = "ward.D2",
scale ="row",
border_color = NA,
annotation_row = labels,
annotation_col = labels,
annotation_colors = ann_colors,
col = colors,
main = "D62 Distances normalized log2 counts")
PCA and MDS
# PCA
gpca <- glmpca(t(cpm.sel.trans), L = 2)
gpca.dat <- gpca$factors
gpca.dat$CellLine <- SampleInfo$CellLine
gpca.dat$gRNA <- SampleInfo$gRNA
gpca.dat$KO<- SampleInfo$KO
gpca.dat$DIFF <- SampleInfo$DIFF
gpca.dat$RAPA<- SampleInfo$RAPA
gpca.dat$Growth_cond = paste(SampleInfo$DIFF, SampleInfo$RAPA, sep="_")
rownames(gpca.dat) = SampleInfo$labels
mds = as.data.frame(SampleInfo) %>% cbind(cmdscale(distance))
mds$Growth_cond = paste(SampleInfo$DIFF, SampleInfo$RAPA, sep="_")
save(mds, gpca.dat, file=paste0(home, "/output/D62_mdsplots.RData"))
ggplot(gpca.dat, aes(x = dim1, y = dim2, color = gRNA,
shape = Growth_cond)) +
geom_point(size = 2) + ggtitle("PCA with log2 counts D62")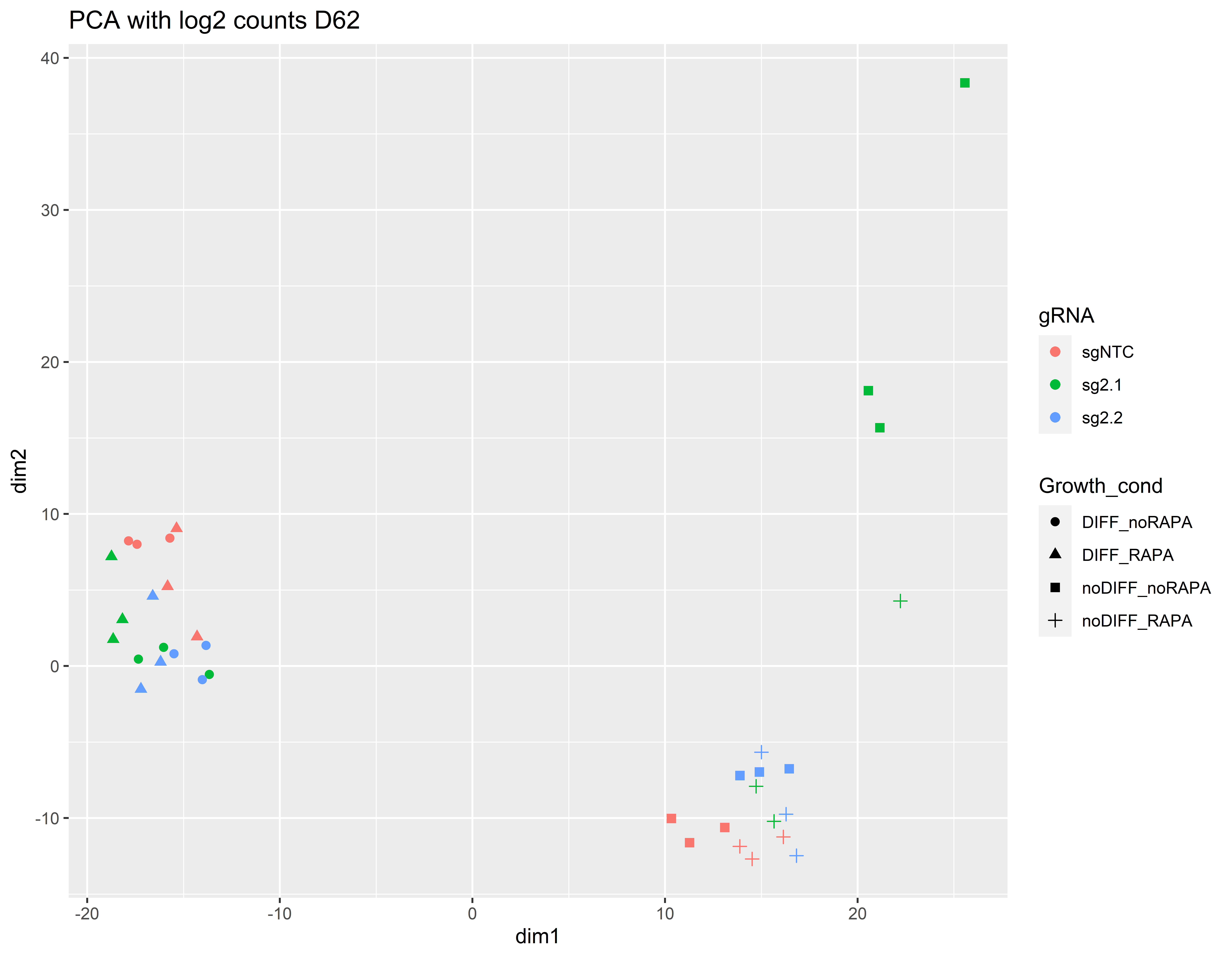
ggplot(mds, aes(x = `1`, y = `2`, color = gRNA, shape = Growth_cond)) +
geom_point(size = 2) + ggtitle("MDS with log2 counts D62")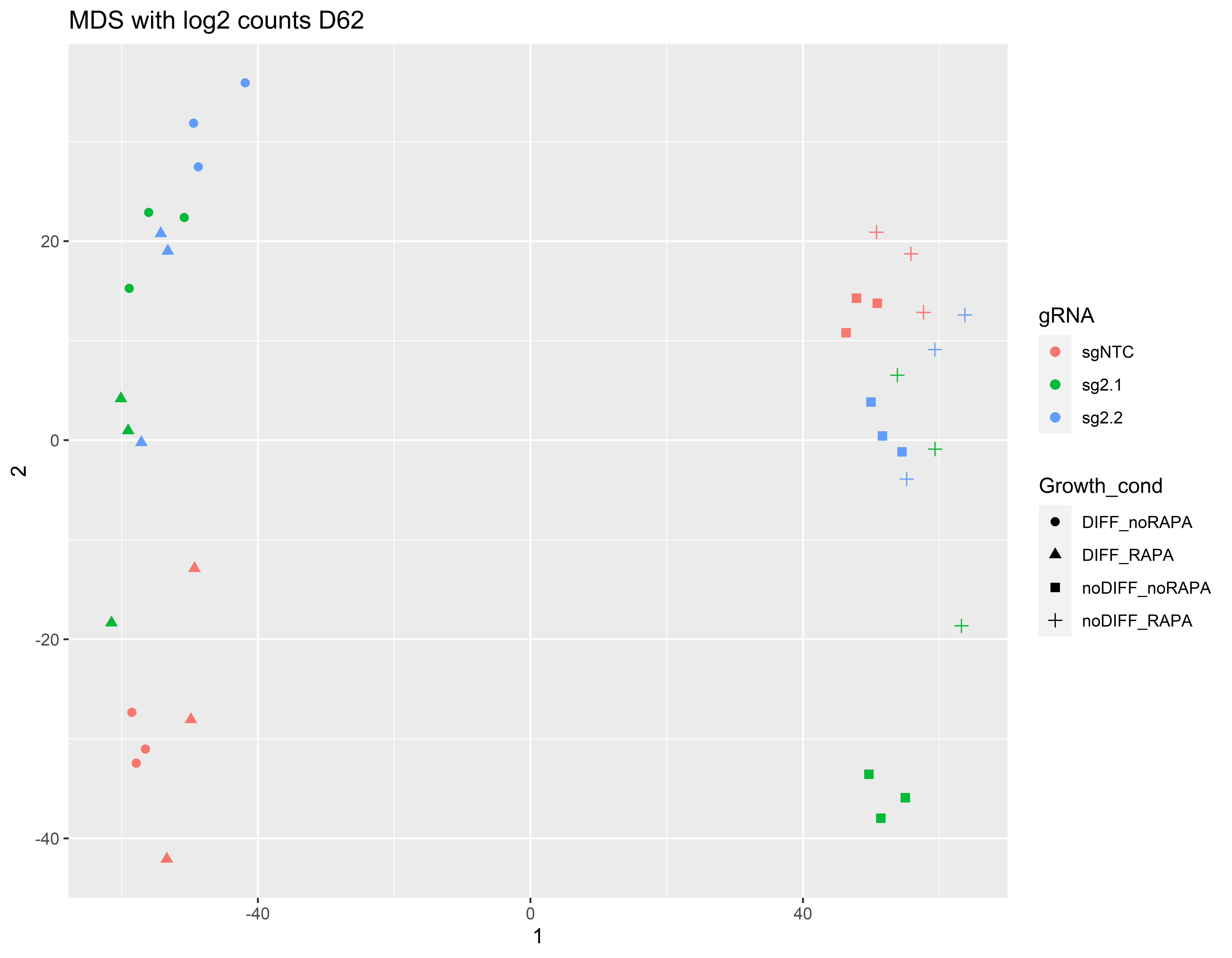
save(ddsMat, file=paste0(output,"/D62_dds_matrix.RData"))
sessionInfo()R version 4.2.0 (2022-04-22 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19045)
Matrix products: default
locale:
[1] LC_COLLATE=German_Germany.utf8 LC_CTYPE=German_Germany.utf8
[3] LC_MONETARY=German_Germany.utf8 LC_NUMERIC=C
[5] LC_TIME=German_Germany.utf8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] compareGroups_4.5.1 kableExtra_1.3.4
[3] gprofiler2_0.2.1 dendextend_1.16.0
[5] pheatmap_1.0.12 forcats_1.0.0
[7] stringr_1.5.0 dplyr_1.1.0
[9] purrr_1.0.1 readr_2.1.3
[11] tidyr_1.3.0 tibble_3.1.8
[13] tidyverse_1.3.2 ggplot2_3.4.0
[15] glmpca_0.2.0 RColorBrewer_1.1-3
[17] DESeq2_1.36.0 SummarizedExperiment_1.26.1
[19] Biobase_2.56.0 MatrixGenerics_1.8.1
[21] matrixStats_0.63.0 GenomicRanges_1.48.0
[23] GenomeInfoDb_1.32.4 IRanges_2.30.1
[25] S4Vectors_0.34.0 BiocGenerics_0.42.0
[27] RCurl_1.98-1.8 knitr_1.42
[29] workflowr_1.7.0
loaded via a namespace (and not attached):
[1] readxl_1.4.1 uuid_1.1-0 backports_1.4.1
[4] systemfonts_1.0.4 lazyeval_0.2.2 splines_4.2.0
[7] BiocParallel_1.30.3 digest_0.6.31 htmltools_0.5.4
[10] viridis_0.6.2 fansi_1.0.4 magrittr_2.0.3
[13] Rsolnp_1.16 memoise_2.0.1 googlesheets4_1.0.1
[16] tzdb_0.3.0 Biostrings_2.64.1 annotate_1.74.0
[19] modelr_0.1.10 officer_0.4.4 svglite_2.1.1
[22] timechange_0.2.0 colorspace_2.1-0 blob_1.2.3
[25] rvest_1.0.3 haven_2.5.1 xfun_0.36
[28] callr_3.7.3 crayon_1.5.2 jsonlite_1.8.4
[31] genefilter_1.78.0 survival_3.4-0 glue_1.6.2
[34] gtable_0.3.1 gargle_1.3.0 zlibbioc_1.42.0
[37] XVector_0.36.0 webshot_0.5.3 DelayedArray_0.22.0
[40] scales_1.2.1 DBI_1.1.3 Rcpp_1.0.10
[43] viridisLite_0.4.1 xtable_1.8-4 bit_4.0.5
[46] truncnorm_1.0-8 htmlwidgets_1.6.1 httr_1.4.4
[49] ellipsis_0.3.2 mice_3.14.0 farver_2.1.1
[52] pkgconfig_2.0.3 XML_3.99-0.10 nnet_7.3-17
[55] sass_0.4.5 dbplyr_2.3.0 locfit_1.5-9.6
[58] utf8_1.2.2 labeling_0.4.2 tidyselect_1.2.0
[61] rlang_1.0.6 later_1.3.0 AnnotationDbi_1.58.0
[64] munsell_0.5.0 cellranger_1.1.0 tools_4.2.0
[67] cachem_1.0.6 cli_3.4.1 generics_0.1.3
[70] RSQLite_2.2.20 broom_1.0.3 evaluate_0.20
[73] fastmap_1.1.0 yaml_2.3.7 processx_3.7.0
[76] bit64_4.0.5 fs_1.6.0 zip_2.2.2
[79] KEGGREST_1.36.3 whisker_0.4.1 xml2_1.3.3
[82] compiler_4.2.0 rstudioapi_0.14 plotly_4.10.1
[85] png_0.1-7 reprex_2.0.2 geneplotter_1.74.0
[88] bslib_0.4.2 stringi_1.7.12 HardyWeinberg_1.7.5
[91] highr_0.10 ps_1.7.1 gdtools_0.2.4
[94] lattice_0.20-45 Matrix_1.5-1 vctrs_0.5.2
[97] pillar_1.8.1 lifecycle_1.0.3 jquerylib_0.1.4
[100] flextable_0.8.1 data.table_1.14.6 bitops_1.0-7
[103] httpuv_1.6.8 R6_2.5.1 promises_1.2.0.1
[106] gridExtra_2.3 writexl_1.4.0 codetools_0.2-18
[109] MASS_7.3-58.1 assertthat_0.2.1 chron_2.3-57
[112] rprojroot_2.0.3 withr_2.5.0 GenomeInfoDbData_1.2.8
[115] parallel_4.2.0 hms_1.1.2 grid_4.2.0
[118] rmarkdown_2.20 googledrive_2.0.0 git2r_0.30.1
[121] getPass_0.2-2 base64enc_0.1-3 lubridate_1.9.1